A chloroplast enzyme safeguards plants against pathological protein aggregation that causes Huntington’s and other neurodegenerative diseases / new research reported in “Nature Aging” may have found a way to “copy” the mechanism for application in human cells
Researchers at the University of Cologne’s CECAD Cluster of Excellence for Aging Research and the CEPLAS Cluster of Excellence for Plant Sciences have found a promising synthetic plant biology approach for the development of a therapy to treat human neurodegenerative diseases, especially Huntington’s disease. In their publication “In-planta expression of human polyQ-expanded huntingtin fragment reveals mechanisms to prevent disease-related protein aggregation” in Nature Aging, they showed that a synthetic enzyme derived from plants – stromal processing peptidase (SPP) – reduces the clumping of proteins responsible for the pathological changes in models of Huntington’s disease in human cells and the nematode Caenorhabditis elegans.
Huntington’s disease is among the so called polyglutamine (polyQ) diseases, a group of neurodegenerative disorders caused by multiple repetitions of glutamine amino acids in specific proteins. An excessive number of polyQ repeats can cause proteins to aggregate or accumulate in harmful and damaging protein deposits, leading to cellular dysfunction and death. To date, nine polyQ disorders have been described in humans. They all remain incurable. Among them, Huntington’s disease is an inherited condition that causes widespread deterioration in the brain and disrupts thinking, behavior, emotion and movement.
Plants are immune to harmful protein aggregation
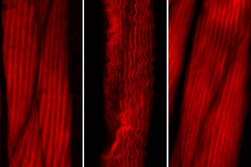
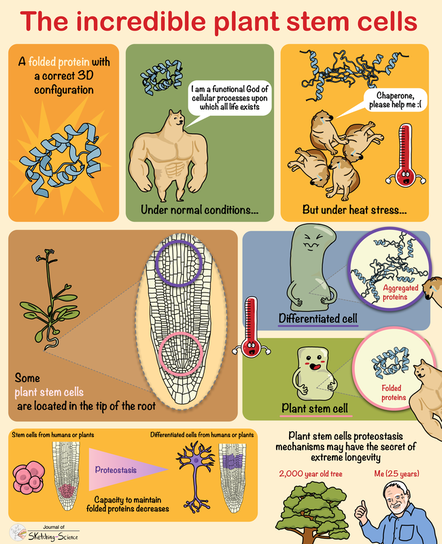
In their recent study, Professor Dr David Vilchez (CECAD) and Dr Ernesto Llamas (CEPLAS) followed an unconventional approach to find potential drugs to treat polyQ diseases like Huntington’s. Plants are constantly challenged by the environment, but they cannot move to escape from these conditions. However, plants possess a striking resilience to stress that allows them to live long. Unlike humans who suffer from proteinopathies caused by the toxic aggregation or cluster of proteins, plants do not experience these kinds of diseases. They express hundreds of proteins containing polyQ repeats, but no pathologies from these factors have been reported. To explore how plants deal with toxic protein aggregation, Dr Ernesto Llamas, first author of the study, and colleagues introduced the toxic mutant protein huntingtin in plants, which causes cell death in human neurons. In contrast to animal and human models, they found that Arabidopsis thaliana plants actively removed huntingtin protein clumps and avoid harmful effects.
By means of synthetic biology, the scientists then transferred the plants’ ability to avoid aggregation into human cultivated cells and animal models of Huntington’s disease. Their hope is that the use of plant proteins could lead to new therapeutic approaches for treating Huntington’s disease and other neurodegenerative diseases.
“We were surprised to see plants completely healthy, even though they were genetically producing the toxic human protein. The expression of mutant huntingtin in other models of research like human cultured cells, mice and nematode worms induce detrimental effects and symptoms of disease,” said David Vilchez.
Plant protein alleviates symptoms in human cells and nematodes
The next step was to discover how plants avoided the toxic aggregation of mutant huntingtin. Indeed, the scientists discovered that the chloroplasts, the plant-specific organelles that perform photosynthesis, were the reason why plants do not show toxic protein deposits. Llamas said: “Unlike humans, plants have chloroplasts, an extra cellular type of organelle that could provide an expanded molecular machinery to get rid of toxic protein aggregates.”
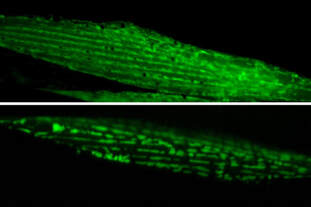
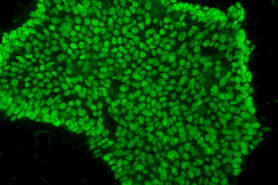
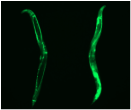
The multidisciplinary team identified the chloroplast plant protein SPP as the reason why plants are unaffected by the problematic human protein. Producing the plant SPP in models of Huntington’s disease such as human cultured cells and worms like the nematode C. elegans reduced protein clumps and symptoms of disease. “We were pleased to observe that expression of the plant SPP protein improved motility of C. elegans worms affected by huntingtin even at later aging stages where the symptoms are even worse,” said Dr Hyun Ju Lee, a postdoc also involved in the study. The results thus open the door to testing SPP as a potential therapy for Huntington’s disease.
Plants as models for aging research
Llamas is convinced that plant research can make a meaningful contribution to treating human diseases. “Many people don’t notice that plants can persist amongst variable and extreme environmental conditions that cause protein aggregation. I believe that plant molecular mechanisms hold the key to discovering new drugs that can prevent human diseases. We usually forget that some plants can live thousands of years and should be studied as models of aging research.” Dr Seda Koyuncu, another postdoc involved in the study, added: “Over the past years, we have seen several promising approaches to treating hereditary diseases like Huntington’s fail. We are confident that our plant synthetic approach will lead to significant advances in the field.”
The team has since acquired funding form the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung – BMBF) through the GO-Bio initial program. “We want to bring our idea into an application. Our plan is to found a start-up to produce plant-derived therapeutic proteins and to test them as potential therapeutics to treat neurodegenerative diseases in humans,” said Llamas.
The research was conducted at the University of Cologne’s CECAD Cluster of Excellence in Aging Research and CEPLAS Cluster of Excellence on Plant Sciences.
Researchers at the University of Cologne’s CECAD Cluster of Excellence for Aging Research and the CEPLAS Cluster of Excellence for Plant Sciences have found a promising synthetic plant biology approach for the development of a therapy to treat human neurodegenerative diseases, especially Huntington’s disease. In their publication “In-planta expression of human polyQ-expanded huntingtin fragment reveals mechanisms to prevent disease-related protein aggregation” in Nature Aging, they showed that a synthetic enzyme derived from plants – stromal processing peptidase (SPP) – reduces the clumping of proteins responsible for the pathological changes in models of Huntington’s disease in human cells and the nematode Caenorhabditis elegans.
Huntington’s disease is among the so called polyglutamine (polyQ) diseases, a group of neurodegenerative disorders caused by multiple repetitions of glutamine amino acids in specific proteins. An excessive number of polyQ repeats can cause proteins to aggregate or accumulate in harmful and damaging protein deposits, leading to cellular dysfunction and death. To date, nine polyQ disorders have been described in humans. They all remain incurable. Among them, Huntington’s disease is an inherited condition that causes widespread deterioration in the brain and disrupts thinking, behavior, emotion and movement.
Plants are immune to harmful protein aggregation
In their recent study, Professor Dr David Vilchez (CECAD) and Dr Ernesto Llamas (CEPLAS) followed an unconventional approach to find potential drugs to treat polyQ diseases like Huntington’s. Plants are constantly challenged by the environment, but they cannot move to escape from these conditions. However, plants possess a striking resilience to stress that allows them to live long. Unlike humans who suffer from proteinopathies caused by the toxic aggregation or cluster of proteins, plants do not experience these kinds of diseases. They express hundreds of proteins containing polyQ repeats, but no pathologies from these factors have been reported. To explore how plants deal with toxic protein aggregation, Dr Ernesto Llamas, first author of the study, and colleagues introduced the toxic mutant protein huntingtin in plants, which causes cell death in human neurons. In contrast to animal and human models, they found that Arabidopsis thaliana plants actively removed huntingtin protein clumps and avoid harmful effects.
By means of synthetic biology, the scientists then transferred the plants’ ability to avoid aggregation into human cultivated cells and animal models of Huntington’s disease. Their hope is that the use of plant proteins could lead to new therapeutic approaches for treating Huntington’s disease and other neurodegenerative diseases.
“We were surprised to see plants completely healthy, even though they were genetically producing the toxic human protein. The expression of mutant huntingtin in other models of research like human cultured cells, mice and nematode worms induce detrimental effects and symptoms of disease,” said David Vilchez.
Plant protein alleviates symptoms in human cells and nematodes
The next step was to discover how plants avoided the toxic aggregation of mutant huntingtin. Indeed, the scientists discovered that the chloroplasts, the plant-specific organelles that perform photosynthesis, were the reason why plants do not show toxic protein deposits. Llamas said: “Unlike humans, plants have chloroplasts, an extra cellular type of organelle that could provide an expanded molecular machinery to get rid of toxic protein aggregates.”
The multidisciplinary team identified the chloroplast plant protein SPP as the reason why plants are unaffected by the problematic human protein. Producing the plant SPP in models of Huntington’s disease such as human cultured cells and worms like the nematode C. elegans reduced protein clumps and symptoms of disease. “We were pleased to observe that expression of the plant SPP protein improved motility of C. elegans worms affected by huntingtin even at later aging stages where the symptoms are even worse,” said Dr Hyun Ju Lee, a postdoc also involved in the study. The results thus open the door to testing SPP as a potential therapy for Huntington’s disease.
Plants as models for aging research
Llamas is convinced that plant research can make a meaningful contribution to treating human diseases. “Many people don’t notice that plants can persist amongst variable and extreme environmental conditions that cause protein aggregation. I believe that plant molecular mechanisms hold the key to discovering new drugs that can prevent human diseases. We usually forget that some plants can live thousands of years and should be studied as models of aging research.” Dr Seda Koyuncu, another postdoc involved in the study, added: “Over the past years, we have seen several promising approaches to treating hereditary diseases like Huntington’s fail. We are confident that our plant synthetic approach will lead to significant advances in the field.”
The team has since acquired funding form the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung – BMBF) through the GO-Bio initial program. “We want to bring our idea into an application. Our plan is to found a start-up to produce plant-derived therapeutic proteins and to test them as potential therapeutics to treat neurodegenerative diseases in humans,” said Llamas.
The research was conducted at the University of Cologne’s CECAD Cluster of Excellence in Aging Research and CEPLAS Cluster of Excellence on Plant Sciences.

 RSS Feed
RSS Feed